|
Even with the 2002 syllabus cuts, this is still a large module and probably will require every bit of the allotted seven weeks for its completion. Despite the rather grandiose title, the module is basically
The ammonia synthesis is used to develop further the students' understanding of chemical equilibrium (which was introduced in the Preliminary Course via precipitation and dissolution of salts, extended to include Le Chatelier's principle in HSC Module 2 (solubility of CO2) and which is now used to optimise yield). The treatment of anion and cation analysis is traditional test tube chemistry and gravimetric analysis. Both are very good for applying basic chemical concepts, though they are not widely used for practical analysis today. The introduction of atomic absorption spectrometry does allow some modernising of analysis. Apart from requiring students to list the main air pollutants and their sources, the syllabus treats in depth only one aspect of air pollution, namely the ozone hole. It uses ozone and CFCs as vehicles for discussing the chemistry of oxygen, ozone and the oxygen atom, and structure and isomerism of haloalkanes. The treatment of water pollution is more systematic, requiring tests for many aspects of water quality and some discussion of several forms of water pollution such as BOD, heavy metal pollution and eutrophication.
Chapter 6 treats Topics 1, 2 and 3 of the syllabus more or less in syllabus order. In the ammonia synthesis the sub-section Source of reactants on pages 193-5 is probably not required by the syllabus. It is included to show (a) that quite a lot of chemistry is involved in preparing a suitable economical mixture of hydrogen and nitrogen for the Haber process, (b) that electrolysis is not the common source of hydrogen, and (c) that air cannot be used as the source of nitrogen unless oxygen is removed first (because of the explosive reaction between hydrogen and oxygen). The sections on analysing mixtures of cations and anions may be more than the syllabus intended, depending upon how you interpret dot point 1 of Section 9.4.3, 'deduce the ions present in a sample from the results of tests'. CC interprets this as meaning several ions present in a sample that is a mixture and hence Sections 6.9 Identifying cations in mixtures and Section 6.11 Identifying anions in mixtures. However if you interpret the ions present as meaning the one cation and the one anion present in a sample, that is a pure substance (or a solution of one substance), then Sections 6.9 and 6.11 are outside the syllabus. The only quantitative analysis left in the Students do column of the syllabus, sulfate analysis of a fertiliser, is treated as a worked example. I had thought with the elimination of all the other specific analyses mentioned in the 2000 syllabus that back titration would not be needed. I can find nothing in the current syllabus document to imply that it is required. Nevertheless the 2005 HSC exam included a question involving a back titration. Hence there is a section below treating it – virtually restoring the bit that had been deleted from the third edition! Chapter 7 on air pollution follows the syllabus sequence very closely. The discussion of photochemical smog is probably outside syllabus requirements: perhaps all that is really necessary is to know that ozone is an air pollutant in the lower atmosphere, not how it gets there. Chapter 8, Monitoring water quality, contains all syllabus material – probably more than is required post-Sept 2002. However there is significant deviation from syllabus order (in order to present a logical development of the various topics). The treatment of the oxygen sensor and total dissolved salts by conductivity and possibly the detection of specific heavy metals (Table 8.3) are probably not required by the syllabus. The discussion of salinity on pages 268–9 probably goes beyond syllabus requirements, but the issue is of considerable interest and concern to Australia at the moment. As with previous modules the
exact relation between Conquering Chemistry and the syllabus for this
module can be seen
in the tables on pages 304–8. Although the syllabus does not specifically say that students should be able to apply Le Chatelier's principle to situations other than ammonia synthesis or CO2 dissolution, Conquering Chemistry assumes that students should be able to work the same sorts of exercises as in the previous syllabus. This is the reason for Exercises 3 to 7 on pages 205–6. Change in number of moles and pressure. In
the absence of any discussion of the gas laws, it is necessary to explain
carefully the relation between number of moles of gas and total pressure: A useful summary of the qualitative features of chemical equilibrium is on page 315–6; the photo on page 316 is particularly informative for adding or removing a reactant. The photos on page 329 (in the Industrial
Chemistry option) are equally relevant to this module. 2. Qualitative analysis of anions and cations It should be stressed that the tests commonly used to identify ions are consequences of the solubility rules of Table 8.1 on page 205 of CCPC (reproduced inside the back cover of CCHSC). Although the syllabus suggests that students do not need to memorise these rules, there are great advantages in doing so. Having to flip back to such a table every time a test is described makes for very slow progress in problem solving. Students also need to be careful whether a particular test exclusively identifies the ion present or whether it just indicates one of several possibilities. For example getting a precipitate with sulfate does not prove that the solution contains lead ions: it could also contain barium, calcium or silver ions (though surprisingly the silver ion is not on the list for consideration). Getting a precipitate with iodide would eliminate barium and calcium. Although a systematic approach to identifying ions is used in CCHSC (using HCl, H2SO4, NaOH in that order for cations, HNO3, Ba2+ in acid solution, Ag+, then Ba2+ in alkaline solution for anions), tests can be done in a variety of different sequences, particularly if only one cation or anion is present. Hence students need to develop skill in interpreting results. Practice on many different exercises is the best way of developing this skill. Although hydrogen sulfide has
traditionally been used in the identification of cations CCHSC deliberately
avoids it, because of both the unpleasant smell and high toxicity of hydrogen
sulfide. Sodium sulfide is reluctantly used in Chapter 8 for the detection of
heavy metals in environmental water. 3. Quantitative analysis (sulfate in fertiliser) The present module requires students to perform just one quantitative analysis, namely sulfate in fertiliser. This introduces students to gravimetric analysis and is the only place gravimetric analysis is treated (apart from the analysis of mixtures in the early part of the preliminary course). The design and performance of gravimetric analyses require care to avoid
These are the sorts of issues
students need to discuss to 'evaluate the reliability of results ... and to
propose solutions to problems ...' (Section 9.4.3 RC DP 4). With the five analyses listed in Section 9.4.3, RC DP 3 of the original (1999) syllabus, it was fairly clear that back titration was required (it's virtually impossible to determine nitrogen or ammonia without it). However when four of these analyses (including the nitrogen analysis) were deleted in 2002, there was no longer any indication in the syllabus document that back titration was required. Hence I deleted it from CCHSC. However there was a question involving it in the 2005 HSC exam paper! It seems that students do need to be aware of the technique of back titration and to work some exercises (an example of syllabus creep that used to plague the pre-2000 syllabus?). Hence the following: Direct titrations as used in Sections 5.8 to 5.12 on pages 157–65 work well if the reaction between the substance to be analysed and the reactant in the burette (the titrant) occurs rapidly. If the reaction is slow because it needs heating or if the substance being titrated is present as a solid, then in a direct titration the endpoint can easily be overshot. A way of avoiding this is to use what is called a back titration. A known excess of a reagent is added to the sample to be analysed, sufficient time or heat is provided for the reaction to go to completion and then the amount of excess is determined by a direct titration with some other solution. The following example illustrates.
Some exercises involving back titration are included in the set for pages 223–4 in the Further Exercises section. Table 7.2 on page 238 lists the main air pollutants and their sources. Some of these (CO2, SO2 and NO2) were also discussed on pages 121–6. Pollution from burning fossil fuels on pages 286–8 of CCPC should be re-visited while studying this section. The main sources of the major air pollutants listed in Table 7.2 on page 238 are:
The operation of catalytic
converters in motor car exhausts is described on page 240–1. Such catalysts were
also mentioned on pages 297 of CCPC where there is a photo of one. All the
commercial catalytic converters in use today are dual bed ones, meaning that
there are small
particles of both platinum and rhodium deposited on the surface
of the ceramic support. Such
catalytic converters for removing NO, CO and hydrocarbons are (confusingly)
referred to as both dual bed (two catalysts) and three-way converters (three types of
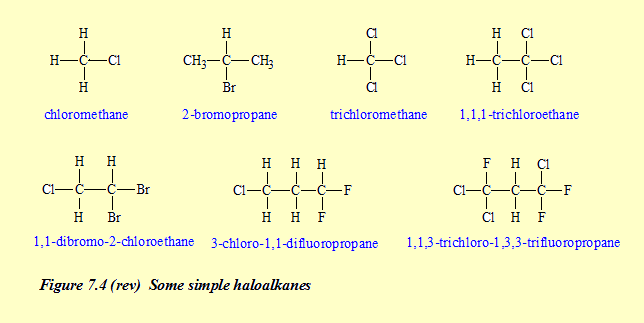
compounds removed). 6. Naming haloalkanes – a bit of a mess! There are problems with this topic on pages 250-1. First, Rule 5 is wrong! The correct rule is given on the errors page, namely
This mistake means that several names for compounds are wrong on pages 249-52 and in some answers to exercises (pages 563-4). These errors are duly noted on the errors page. (Click here to go there.) These corrections have been made on pages 249-52 of the electronic version of the book on the NelsonNetBook website (available with copies of the book purchased since 2013) but it was not possible to correct the wrong answers on pages 563-4 (Click here to go to the corrections for those pages.) The second problem is that Rule 3 is expressed differently from the official IUPAC rule, though the rule on page 250 leads to the correct name for compounds up to eight carbon atoms (that is, up to octanes) which is as far as you have to go for the HSC – and the page 250 rule is probably easier to understand and apply. Following the correct IUPAC method, Rule 3 should be:
Using the correct method for Rule 3 does not lead to any consequential changes in the following pages of CCHSC. Matters came to a head with Question 12 in the 2012 HSC exam paper (correct name for a haloalkane). Assuming no error in CCHSC quickly led students to the correct answer, D. However students who knew ot the error in CCHSC were left with no correct answer for the question, though three of the offered answers could be easily eliminated leaving only D. (See Answers for the 2012 exam paper.) The Board of Studies drew teachers attention to this matter in an Official Notice dated 1 March 2013 and referred them to a Royal Australian Chemical Institute (RACI) document on naming organic compounds (http://www.raci.org.au/document/item/1012). However there are some problems with that document also! The RACI document interprets the mention of 'isomers' in Dot Point 9 of Section 9.4.4 of the HSC syllabus as including branched-chain haloalkanes, not just straight-chain ones, and so its rules become more complicated because they take into account the possibility of alkyl substituents (side groups) as well as halo ones. The document uses both the terms, 'substituent' and 'functional group'. Generally in chemistry these two terms have different meanings. However in the context of naming haloalkanes there is no distinction between these two terms; alkyl groups are treated identically to halo atoms. The NB sentence in Step 5 is the hardest to interpret and its interpretation is not helped by their Example 3, because it is Step 6 that decides the name there, not Step 5 as stated. An illustration of the use of Step 5 is the compound Br–CH2–CH2–CHCl2. This is either (a) 1-bromo-3,3-dichloropropane (numbering from the left) or (b) 3-bromo-1,1-dichloropropane (numbering from the right). The two sets of numbers (locants) listed in ascending order are 133 for (a) and 113 for (b). (b) has the smallest number (1) at the first point of difference (second number) so the correct name is (b) 3-bromo-1,1-dichloropropane. The RACI Chemical Education Group is expected to revise this document early in 2014. CCHSC interprets 'isomers' in that Dot Point 9 as meaning just different positioning of halo atoms on straight-chain compounds as on page 250 and in exercises on pages 251-2. It adopts this interpretation because in all other classes of carbon compounds treated, the syllabus specifically refers to straight-chain ones only. Perhaps after all this it would be worthwhile to set out the steps for naming haloalkanes in their entirety without any errors.
7. Ozone destruction in the stratosphere It is essential to appreciate that one chlorine atom can destroy thousands of ozone molecules (in what we call a chain reaction): this is why very small concentrations of CFCs in the atmosphere can destroy a significant proportion of the ozone in the stratosphere. It is also important to
appreciate that Cl + O3
® ClO + O2 and ClO +
O ® Cl
+ O2 alone do not explain the ozone hole: they explain a general
reduction of ozone throughout the whole stratosphere. It is the liberation of Cl2
from HCl + ClONO2
and the subsequent formation of extra Cl atoms that cause the dramatic reduction
in ozone concentrations that are observed over the Antarctic in spring (pages
253–6). 8. Water quality criteria and types of water pollution Page 263 lists eleven criteria for assessing water quality. Perhaps we could add to this list colour and smell (though these are consequences of the presence of various organic compounds, organisms or metal ions). Students probably need to memorise rough values of several of these criteria for clean water and how the values for polluted water vary (higher or lower) from these – such as for TDS. pH, DO, BOD and faecal coliforms. See for example Table 8.1 on page 264. The major types of water pollution and their main sources are:
Or presenting some of this information in a different way, the major sources of water pollution are
As in previous modules most of the material in this section is not required by the HSC syllabus. It is presented here either because it provides better understanding of syllabus material or because of its inherent interest to HSC students. 1. Colorimeter
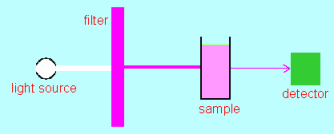
The photo shows a commercial colorimeter. The white square at the top right of the instrument is the cap of the cell that is being measured. Typical cells containing other samples for measurement are on the left.
A spectrophotometer or spectrometer is a much more sophisticated (and expensive) instrument than a colorimeter: A spectrometer uses a monochromator which is able to select a much narrower wavelength slice of light to shine through the sample (a few nanometers instead of a 100 or so nanometers), is able to measure the reference and sample simultaneously and is able to scan through a whole wavelength range (e.g. from 200 to 800 nanometers) and record absorbance as a function of wavelength. A colorimeter is essential for the analysis of phosphate
( and
nitrate) in environmental water (pages 280–1) but not for phosphate
in washing powders or fertiliser (page 223–4) because its concentrations there
are so much higher. 2. Some history of concerns
about stratospheric ozone In 1968 Harold Johnston of the University of
California, Berkeley, presented arguments to show that nitrogen oxides (NO and
NO2) in the exhausts of high-flying aircraft would destroy
stratospheric ozone. He suggested that a fleet of 1000 supersonic commercial
aircraft flying just above the tropopause would destroy 10 to 20% of
stratospheric ozone. The chemical reactions he proposed were similar to the
chlorine atom reactions discussed earlier with NO taking the place of the Cl: At that time both Russia and an Anglo-French consortium were building supersonic commercial jet aircraft, while the USA, having put its efforts into developing "jumbo" sub-sonic aeroplanes, was considerably behind in the development of commercial supersonic planes. Concerns that were a mixture of environmental, commercial and political led to President Nixon setting up the Climatic Impact Assessment Program (CIAP) in the early 1970s to investigate the effects of commercial supersonic flights on the environment (particularly on the stratosphere). CIAP funded a lot of scientific research into the chemistry and physics of the stratosphere over the next 15 years. However before CIAP could complete its report two things happened. First the world suffered a major energy crisis in 1973-4 when the oil-producing countries turned off the supply of oil in response to an Arab-Israeli war. This dramatically increased awareness of the need for energy efficiency and this was a cold blanket for supersonic flight (which used about four times the amount of fuel per passenger mile as does sub-sonic flight). This and other environmental (political?) issues virtually killed supersonic passenger travel except for limited use of the Anglo-French Concorde (which still operates though roughly to the same limited extent as it did in the mid-1970s). The second event was a claim by Molina and Rowland in 1974 that CFCs were a greater threat to stratospheric ozone than oxides of nitrogen. They produced figures on the amounts of CFCs released into the atmosphere and on the present and predicted rates of growth of those amounts and described the chemistry that could lead to ozone destruction. This caused the CIAP scientists to divert the emphasis of their studies to the effects of chlorine. All of the studies on ozone depletion during this time focused on the whole global stratosphere; it was not envisaged that there could be a localised stratospheric problem. Attention concentrated on the chemistry occurring in the gas phase; little thought was given to the possibility of significant reactions occurring on the surfaces of solid particles (of which there are very few in the stratosphere, considered globally). A final consensus report on all of this work was published in 1984. It concluded that both CFCs and oxides of nitrogen posed some threat to stratospheric ozone, but not nearly as great as had been first suggested. It concluded that the then current levels of CFCs and a proposed small fleet of supersonic transports would deplete stratospheric ozone by about 5 to 15% (depending upon which of the several computer models was used). It was in response to this report that the international conference that lead to the Montreal Protocol was called. However the report was out of date before the conference could meet! The ozone hole – a far more serious problem – was reported in 1985. The original Montreal Protocol was developed to deal with the expected 5 to 15% decrease in stratospheric ozone. The much greater decrease in ozone over the Antarctic and the further "deepening" of the ozone hole in subsequent years required the two revisions to the Montreal Protocol that have occurred. After the original discovery of the ozone hole in
1985 there was a massive concerted effort by scientists worldwide to confirm the
existence of the hole, to map its extent year by year and to work out what was
causing it. 3. Conferences on ozone hole
and greenhouse Conferences on the ozone hole have produced restrictions on the emission (and production) of CFCs for the purpose of minimising the ozone hole. Such restrictions do have minor benefits for the greenhouse problem but conferences on greenhouse have not specifically targeted CFCs. Ozone is also a greenhouse gas so its formation in the lower atmosphere could be of concern. However ozone is formed only in large cities with high intensities of sunlight, and while this is a problem in those localised areas, the ozone is soon dispersed and destroyed so makes only a negligibly small contribution to global warming. Ozone hole conferences
However increasing concern over the ozone hole made it clear that these steps were not sufficient to control the problem. Hence there was a further agreement in London in 1990 to
With the continued worsening of the ozone hole, pressure continued for a quicker phasing out of CFCs. Hence in Copenhagen in 1992 it was agreed to
The more developed countries (including Australia) that signed these agreements have done a fairly good job in adhering to their terms and timetables. Greenhouse conferences In June 1992 the United Nations Conference on Environment and Development was held in Rio de Janeiro; it is widely referred to as the 1992 Earth Summit. This was the largest gathering of heads of government in history and the most widely publicised conference on the environment ever. It produced, among other equally important documents, the Framework Convention on Climate Change which despite noble sounding sentiments contained no definite timetable for emission reductions. Australia signed this framework agreement. There were follow-up conferences (of little consequence) in Berlin in 1995 and in Geneva in 1996. The first real attempt to introduce specific limits for greenhouse emissions was the Kyoto Conference (in Japan) in December 1997. Details of this agreement are given in the Greenhouse section of the Preliminary Course Module 4 web page. A follow-up conference in the Hague in November 2000 produced little of significance. In Bali in December 2007 there was a conference to discuss strategies for a post Kyoto agreement (beyond 2012). This meeting set an overall guideline of a 25% to 40% cut in emissions by 2020. A conference is scheduled for Copenhagen in 2009 to develop the details of a new agreement. In contrast to agreements on eliminating the ozone hole, conferences on restricting greenhouse emissions have produced very mild proposals for restricting greenhouse emissions (which will have very little effect) and even these have not been agreed to by many countries (notably the US), and apart from the European countries there has been little move toward implementing these weak restrictions. There is little cause for optimism about the greenhouse problem.
040214 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||