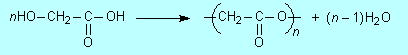Some general commentsThis module draws together some fairly diverse areas of chemistry – hydrocarbon fuels and plastics (polymers), electrochemistry and nuclear chemistry. As teachers you will have to work hard to unify these varied topics. To some extent energy provides a unifying theme – crude oil being processed into various products for particular uses, some non-hydrocarbon sources of energy, electrochemistry providing easily portable energy sources, and nuclear energy, though the syllabus does not actually include nuclear energy despite nuclear reactors being mentioned. The jump from simple addition polymers to condensation polymers and cellulose is a massive one for students whose only previous experience of organic chemistry has been straight-chain hydrocarbons. Care will be needed in introducing this material to avoid confusing or demoralising students. The section of the syllabus on nuclear chemistry seems quite straightforward and short at first reading – but the listed topics require a lot of extra material to make them understandable. While nuclear fission is not mentioned, it has to be described in order to explain how a nuclear reactor works and hence how commercial radioisotopes and transuranic elements are produced. Similarly while alpha, beta and gamma rays are not mentioned, some account of them is necessary to explain the nature of radioactivity and the operation of instruments used to detect radiation. And half-life is probably needed to explain why certain isotopes are used for particular purposes. So overall this short section of the syllabus involves quite a large slab of material. Note that this is the only core module that
addresses HSC Course outcome H5, namely 'identifies possible
future directions of chemical research'. This would appear to be
the main reason for the inclusion of the item in the right-hand column of
Section 9.2.2 of the syllabus document and of the
vanadium cell and the Gratzel cell in Section 9.9.4. Perhaps its also the reason for inclusion of the
completely useless description of how transuranic elements are produced. The syllabus and Conquering ChemistryConquering Chemistry follows syllabus order quite closely as an inspection of the tables on pages 98–101 will show. Syllabus sections 1, 2 and 3 are treated in Chapter 1, Section 4 in Chapter 2 and Section 5 in Chapter 3. Perhaps it can be argued that the reactions in Figure 1.1 on page 9 are sufficient for syllabus requirements; however Section 1.5 has been included to meet the 'transformed into many useful products' of dot point 2 of Section 9.2.1. This section also serves to illustrate that industry frequently uses highly specialised catalytic reactions rather than the 'ordinary' general reactions of the compound involved, primarily because yields are better and rates higher. While Sections 1.6 and 1.7 treat the three addition polymers directly referred to in the syllabus document, CCHSC also includes mention of four other common addition polymers (in Section 1.8). Section 1.9 of CCHSC, Relating properties and uses to structure (of polymers) probably goes into greater depth than is strictly required by the syllabus, but without this depth it is hard to understand why particular polymers are used for particular purposes. Its hard to know just how much detail the syllabus requires for the structure of cellulose at this very early stage in the students' developing knowledge of organic chemistry. If what is on pages 23 and 25 is deemed insufficient, there is more detail on pages 470-3 (in the Forensic Chemistry option). The syllabus does not mention oxidation number, so CCHSC has taken a very simplistic approach to 'oxidation state' (= valence for monatomic ions) on pages 44–5. This is sufficient for systematic naming (iron(III) chloride, copper(I) oxide etc) and for relating changes to electron gain and loss in the redox reactions required in the course. Balancing equations for reactions of permanganate and dichromate are not required so there is no need for a formal treatment of oxidation number. An account of the nickel-cadmium cell has been included (page 59), because that cell is so commonly used today, although the syllabus makes no mention of it. Similarly the silver oxide button cell is discussed (on pages 54–5) though not mentioned in the syllabus. At first sight Chapter 3 on nuclear
chemistry appears to include material that is not specifically
listed in the syllabus (Section 9.2.5). However these non-listed
items do seem to be necessary for proper understanding of topics
that are listed (e.g. nuclear fission for production of
transuranic elements). I have included half-life though it could
be argued that this is not needed (though it is relevant to
choosing isotopes for particular purposes). 1. Naming monomers and polymers There are often arguments among students and teachers about the 'correct' names for several common substances such as ethylene and propylene (and acetic acid and ethyl acetate). This arises because there are 'systematic' names and 'IUPAC preferred' names. The International Union of Pure and Applied Chemistry, IUPAC, is the organisation that decides upon the names for chemical compounds; among other things it formulates rules for the systematic naming of compounds. Despite this, because of long-entrenched usage of trivial names, there are some compounds for which IUPAC has decided that the old trivial names are to be the preferred names. Some of the common ones are:
Naming polymers Simple polymers such as those from the above monomers are generally named by what is called a source-based or monomer-based method. This involves
This method of naming leads to
Confusion over brackets arises partly because there is another system of naming polymers, called a structure-based method. It is preferred by IUPAC particularly for more complex polymers than being treated here. In that method the name of the repeating unit is always put in brackets. Occasionally that method leads to the same name as the monomer-based method, except for the brackets: for example it gives poly(propylene) and poly(tetrafluoroethylene). Note that polythene is the trade name of a
particular brand of polyethylene. Similarly teflon is the trade
name of one company's polytetrafluoroethylene. As teachers we should try to minimise the amount of jargon (and number of initialisms or acronyms) that we use in an effort to keep our subject as intelligible to newcomers as possible. Initialisms for polymers that we probably have to put up with are PVC, PET (and PTFE if we intend introducing polytetrafluoroethylene (teflon)). Perhaps we should resist using HDPE, LDPE and some less commonly used ones. Note that while in polymer contexts PAN is
sometimes used for polyacrylonitrile, PAN is more widely used for
peroxyacyl nitrates (or peroxyacetyl nitrate), components of
photochemical smog. (CCHSC pages 123 and 239–40). Considerable effort is being made today to recycle plastics. There are two reasons for this: first to conserve raw materials (particularly crude oil) and secondly to minimise environmental pollution by plastics which because of their low biodegradability stay in the environment for extremely long times. To aid recycling many articles made of plastic are embossed with a symbol indicating which plastic the article is made of. A typical recycling symbol is shown below; the number inside the triangle identifies the particular plastic (polymer):
Sometimes, but not always, the symbol has the initials of the polymer under the triangle.
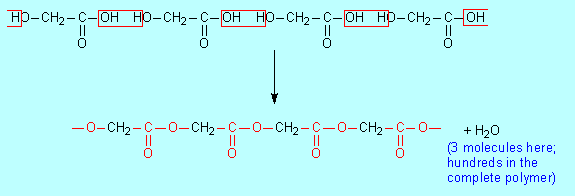
4. Condensation polymers Condensation polymers are introduced in this module via the need to consider cellulose as a future potential source of chemicals currently obtained from petroleum. However this is very early in students development of understanding of organic chemistry and it might be helpful to revisit this material after studying alcohols, acids and esters in Module 2. Many important condensation polymers are polyesters, so discussion of these after learning about esters and their formation makes sense and may make the topic more easily understandable. The simplest polyesters are poly(glycolic acid) and poly(lactic acid), both of which are used in medical sutures that dissolve in the body over time. This means that they can be used in internal stitching where mechanical removal after healing is not practical. The structures of these two acids
are: Because these molecules have an acid group at one end and an alcohol group at the other end they can polymerise by undergoing an esterification. For glycolic acid: The key part of the ester functional group is shown in red. An alternate way of writing this equation is:
Exercise: Write similar equations showing the conversion of lactic acid to poly(lactic acid). The poly(3-hydroxybutanoate) discussed on pages 28–9 is also a polyester. Again the monomer has an OH group at one end and a COOH group at the other and so can undergo esterification. The most widely used polyester is poly(ethylene terephthalate), PET. There are many polyesters, but in everyday language 'polyester' generally means this particular compound, especially when used for fabrics, carpets and textiles generally. This same polyester is widely used to make drink bottles; then it is usually called PET. Although polyester clothing and drink bottles seem very different, they are basically the same chemical substance. Exercise 29 on page 24 asks you to draw the structure of PET which is shown in the answer on page 545. The other major group of synthetic condensation polymers are the nylons. To understand the structure of nylons we need to introduce a class of compounds called amines. R–NH2. Amines react with carboxylic acids to form amides (page 23). There are compounds that contain both an amine group and a carboxylic acid group: they are called amino acids (compare with the hydroxy acids used for the simple polyesters above). The simplest nylon, called nylon-6, was introduced on page 23. It is made from an amino acid, 6-aminohexanoic acid. The commonest nylon, called nylon-66 is made from two monomers, hexanedioic acid (or a chlorine derivative of it) and 1,6-diaminohexane. You are asked to deduce its structure in Exercise E1 below (answer is given). Kevlar, used in bullet-proof vests, is another type of nylon. See Exercise E This same amide linkage occurs in the naturally occurring polymers called proteins, mentioned on page 23 and treated in detail in the Forensic Chemistry option on pages 480–8. Starch and cellulose are other naturally occurring polymers. Both are formed from glucose. However the reaction involved in forming the polymer is not one that we meet in our studies of small molecules in this course. It is however a condensation reaction: a molecule of water is eliminated between each pair of glucose molecules. This polymerisation is treated very simply on pages 22–3 and discussed in more detail in the Forensic Chemistry option on pages 470–2, though to understand the difference between starch and cellulose you need to read pages 467–8 and possibly 474–8.
5. Cellulose, biopolymers, ethanol Section 9.2.2 of the syllabus requires discussion of the possible need for alternative sources of compounds currently obtained from petrochemicals. It suggests that cellulose could be such an alternative source, but without going into specifics, then turns to developments in synthesising biopolymers. Section 9.2.3 then looks at ethanol as an alternative source of chemicals (particularly ethylene) or as a fuel, with the ethanol coming from fermentation of sugars or starch. This source of ethanol or ethylene is industrially well-established, though not always economically advantageous. Note that it is difficult to get ethanol from cellulose, because it is hard to break cellulose into glucose (though good progress has been made recently). The full structure of cellulose is probably a bit indigestible for beginning Year 12 students, so I have given just the bare minimum on pages 23 and 25. There is more detail (including the relation to starch) on pages 470–3. On page 23 of CCHSC the condensation reaction between glucose molecules looks like a reaction between two alcohol groups. If you (teachers) are worried that alcohols do not react with one another, remember that one of the OHs involved is actually a hemi-acetal (derivative of a carbonyl compound) which does react with an ordinary alcoholic OH. A full explanation of this condensation and of the distinction between cellulose and starch really requires some discussion of carbonyl chemistry (aldehydes and ketones). Because this is not in the syllabus, I have tried to skim over it as simply as possible. If you are puzzled about 'the basic carbon-chain structures needed to build petrochemicals' of the fifth dot point on Section 9.2.2 of the syllabus, join the club! The phrase appears to come from a book, ChemCom: chemistry in the community, 2nd edition, published for the American Chemical Society by Kendall/Hunt Publishing Coy, Iowa USA, 1993 (ISBN 084035505X) page 211. However there is so little relevant information in the two-page article that itis not worth tracking down the book. There is no follow-up on what chemicals could be obtained from cellulose, so we are left with just the possibility of ethanol and hence ethylene, i.e. just C–C – not much of a carbon-chain structure!. A good source of information about biopolymers is Barnum, Susan R., Biotechnology – an introduction, 1998. I have used poly(3-hydroxybutanoate), PHB, as the named biopolymer to discuss the industrial production, properties and use of, though there are other biopolymers that could be treated. This is discussed in Barnum. The property of PHB that make it particularly interesting is its biodegradability. Another biopolymer that could be discussed
in this context is EPO, ethryopoietin. It helps the body make extra red blood
cells and medically has potential for reducing the need for blood transfusions. However it
has also been used illicitly by some athletes to enhance performance and was
first routinely tested for during the Sydney Olympics. It is made using enzymes extracted from Chinese
hamster ovary cells. 6. Heat of combustion of alkanols The syllabus requires students to perform an experiment to determine then compare the heats of combustion of three alkanols on a per mole and per gram basis. The usual experiment (burning liquid in a spirit burner to heat water) has large experimental errors so in order to get significant differences between the kilojoule per gram values for different alkanols it is necessary to use alkanols that differ as much as possible in molecular weights – ranging say from methanol to pentanol (hexanol is a bit too viscous and non-volatile to burn well). The differences for alkanols are somewhat larger than for the hydrocarbons (on a per gram basis). The experiment can be extended by adding
some data processing for carbon monoxide and sucrose and
correlating heat released per gram with percentage oxygen in the
compound. 7. The dubious equation ΔH = – m C ΔT This equation is generally given on the HSC exam data sheet without any definition of what the symbols mean, particularly ΔH. ΔH is normally the molar enthalpy change for a chemical reaction and so the equation is only correct for a very specific case – when one mole of a substance undergoes chemical reaction (such as combustion) and heats up a mass m of another substance (such as water) with a specific heat capacity C through a temperature change, ΔT. The equation that is always correct is q = m C ΔT .... (a) where q is the amount of heat absorbed when a mass m of a substance with specific heat capacity C is heated through a temperature change of ΔT. For example if 250 g water of heat capacity 4.2 J g–1 K–1 is heated from 25oC to 65oC, then the amount of heat absorbed (by the water) is q = 250 X 4.2 X (65 –25) g J g–1 K–1 K = 4.2 X 104 J = 42 kJ If this heat had been released from the burning (combustion) of 1.4 g ethanol, then the heat released per mole of ethanol (46 g) would be 42 X 46/1.2 = 1.6 X 103 kJ/mol. ΔH for the combustion reaction is the heat absorbed when the reaction occurs which is minus the heat released so ΔH = –1.6 X 103 kJ/mol. If the temperature falls, that is if ΔT is negative, then the heat absorbed is negative, meaning that heat is actually released. ΔT is always final temperature minus initial temperature If heat is absorbed (ΔT positive), then q is positive; if ΔT is negative, then heat absorbed q is negative; a negative heat absorbed means that heat is actually released. Unfortunately this material is spread out in Conquering Chemistry. The basic equation is introduced and illustrated in CCPC on pp.222-4, then applied to calculating ΔH for a chemical reaction on pp.278-81. Then measuring heat of combustion is treated in CCHSC on pp. 36-7. One
other thing should be noted about equation (a) and that is the units of C.
In the example above the units of C were given as J g–1 K–1.
The units could be given in J kg–1 K–1 or in kJ kg–1
K–1. This means that for water C could be given as 4.2 J g–1
K–1 or as 4.2 x103 J kg–1 K–1 or
as 4.2 kJ kg–1 K–1.
You need to look closely at the units given with the value of C and use
appropriate units for the other quantities and for the answer. The HSC
examiners seem to prefer J kg–1 K–1 but there is no
guarantee that they will continue to do so, so watch your units! 8. Activity series and comparison with table of standard electrode potentials Although the current syllabus does not require students to use the table of half reactions and standard electrode potentials to make predictions about relative reactivity, it may be worth drawing attention to the similarity of this table to the activity series and how it can similarly be used to make predictions about which metal will reduce which metal ion (as they are required to do with the activity series). To do this we note that
Another way of summarising reactivity in terms of the table of electrode potentials is to say that the half reaction with the higher electrode potential has the greater tendency to occur. Because the activity series can be written
across the page (as in CCHSC page 43 and Chemistry Contexts 2 page
63) or down the page (as
in Pathways 1, page 145 or 2 page 52), and electrode
potentials listed in descending order (CCHSC page 67) or
ascending order (Pathways 2 page 62 and Chemistry Contexts 2
page 69), students may need to
be warned that particular statements refer to the series or table
written in a particular way (unless you use a statement that is
format independent as in the statement above). 9. Calculating voltages for electrochemical processes When I first read the Students learn to column of the electrochemistry section of the syllabus, I happily thought we would at last be able to teach electrochemistry without having to bother with calculations using electrode potentials, but they get stuck on at the end of the students do column – and so they get stuck on at the end of Chapter 2 and the treatment is quite brief. I would much prefer to have been able to end this chapter at page 64. There are three types of calculation that students need to be able to do
Perhaps some additional worked examples would be helpful.
We break the reactions into half reactions and obtain their standard EMFs from a table of standard electrode potentials such as on page 67, then use Equation 2.12 on page 68:
For the second reaction:
We use Equation 2.14 on page 70:
1. Some background to current research in energy and related fields The industrial revolution in Britain and Europe in the first half of the nineteenth century led to dramatically increasing use of coal as a fuel (wood had been the major fuel before that). The widespread introduction of electricity in the first decades of the twentieth century saw further increases in coal consumption. Introduction of motor cars resulted in the widespread use of crude oil as a source of fuel (petrol). Exploration for oil led to discoveries of natural gas which was also widely utilised as a fuel. So by the middle of the 20th century, fossil fuels were being used at a huge and ever-increasing rate. In the 1950s and 60s there was a significant movement away from coal towards oil, because coal-burning was causing severe air pollution in large cities: oil-burning power stations were much cleaner than coal-burning ones. At this time nuclear energy for electricity generation was introduced – first because it was cleaner (no severe air pollution) and secondly because it was thought it would be cheaper in the long run (information about long-term storage of wastes and high de-commissioning costs was lacking). In the late 1960s, early 70s a group of scientists and economists known as the Club of Rome first drew attention to the finite fossil fuel resources of the Earth and sounded warnings about their possible exhaustion, particularly of crude oil, the least abundant and most highly used of the fossil fuels. In 1974 as a consequence of the 1973 Arab-Israeli war the Organisation of Oil Producing Countries, OPEC, was formed and for a period cut off oil supplies to the developed nations (USA and Europe in particular). This caused great upheaval to most of the affected economies and had a dramatic impact upon the attitudes of the leaders in those nations towards energy resources. For the first time they began to think in terms of oil supplies running out. This provided impetus for a diverse range of research programs aimed at developing alternative sources of energy. At that time the emphasis was on finding alternative sources of energy: little attention was given to reducing overall energy use. Some of the lines of research that were followed were
Although environmental scientists had been warning about the enhanced greenhouse effect (global warming) due to increased discharges of carbon dioxide into the atmosphere) since the early 1970s, little notice was taken of them in the 1970s and 80s; they could not produce convincing evidence of the effect – just theories. However by the time of the 1992 Rio Earth Summit the evidence for the reality of global warming had become more convincing and the theories about it almost universally accepted by the scientific community. Consequently that conference recognised the seriousness of this problem and proposed future conferences to set limits upon greenhouse gas emissions (the latest ones being in Kyoto in 1997 and Bonn in 2001). Acceptance of the reality of the enhanced greenhouse effect (global warming) has had significant impacts upon energy research. There is now less enthusiasm for developing the alternative fossil fuels of oil shale and tar sands – they will just release more carbon dioxide per kilojoule of energy produced than coal and oil. So too will liquefaction or gasificatioin of coal. Biomass is also proving somewhat disappointing: it will probably never be greenhouse neutral as originally thought – too much energy in the form of fertiliser and processing costs is required – and there is growing recognition that expanded agriculture on marginal land will have severe environmental costs (soil erosion, salinity, stream siltation, fertiliser and pesticide run-offs etc). There is growing recognition that the direct harvesting of solar energy (by the methods outlined above) is about the only major way that future energy needs can be met without further aggravating the enhanced greenhouse effect. Wind and tidal energy are forms of solar energy There is also growing recognition that the world is probably unable to supply sufficient 'greenhouse friendly' energy to meet the current profligate energy use by developed countries and reasonable expectations of developing nations: we will have to learn to live with using less energy and use our energy much more efficiently. This will probably require lifestyle changes for people in developed countries, though this is an aspect of the energy debate that most politicians tend to ignore, because it would be a great dampener on economic growth which these politicians see as essential to the continued improvement in quality of life for their voters, though some people argue that quality of life is not synonymous with living standards or increased consumerism. There is a full discussion of the enhanced greenhouse effect in the Preliminary Course Module 4 page of the ConqChem website. To go to it click on The enhanced greenhouse effect Nuclear energy With growing concerns about global warming, there is now growing thought that maybe nuclear energy with proper safeguards built into it may be part of the answer to the problem of providing energy without greenhouse emissions. However detailed studies are needed on the overall energy efficiency of nuclear reactors: that is, does a nuclear reactor during its lifetime produce more energy than would have been produced by the fossil fuels that were used to generate the nuclear energy – the fossil fuel energy needed to mine and enrich the uranium, to build, operate and maintain the reactor, to store safely for thousands of years the radioactive byproducts of the reactor and to decommission the reactor at the end of its economic lifetime. Despite the lack of clear-cut answers to many of the questions surrounding nuclear energy such as the greenhouse one, safety, sabotage and terrorism, weapons proliferation and long-term storage of radioactive wastes, many countries, including China and India as well as the old nuclear club, are now building or planning to build many new nuclear reactors. The driving forces have been economic ones; the escalation in oil prices in recent years (and the consequent rises in coal prices) also) and the unlikelihood of them ever returning to the levels of five or ten years ago have made nuclear energy appear less expensive than non-nuclear energy at least when the calculations are done on a narrow short-term basis. It is this boom in building nuclear reactors in other countries and the consequent need for uranium (of which Australia is one of few countries to have plentiful supplies) that has triggered the current (2006) debate on nuclear energy in Australia. Relevance
for the HSC If we use up all the Earth's crude oil as fuel for cars, planes and trains, then we will need to find other sources for the raw materials needed to make plastics. Currently we can get ethanol from starch which is a minor constituent of plant material and we can convert ethanol into ethylene and so make many of our current plastics from it. However it would be better if we could use cellulose to make ethanol, because there is far more cellulose in plants than starch. Until recently breaking cellulose into glucose (a required first step before the conversion of glucose to ethanol) has been very difficult and not commercially practical. However recent research is providing ways that may make this practical in the near future. There is an alternative argument (given in CCHSC) that as oil supplies become depleted, price will escalate to such an extent that it will become too expensive to burn and so will still be available for plastics manufacture. However there is another factor associated with developing alternative sources of plastics and that is the ability to build in biodegradability. Hence the segment on poly(3-hydroxybutanoate).
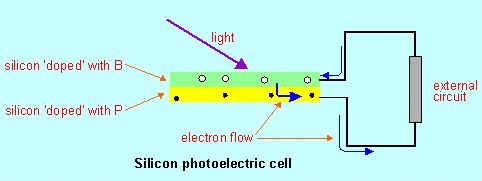
2. Vanadium redox cell and the Gratzel cell I have some misgivings about including these two cells in CCHSC because I think the syllabus really wants students to search out this information for themselves. However in the internet sites I can find on them the chemistry is either difficult to follow or not explained very fully. Hence reluctantly I have decided to include descriptions of the actual chemistry in the book (pages 61–4). Students should seek out other sources of related information, starting with the websites given below. The Gratzel cell (a liquid junction photovoltaic device) is also called a photo-sensitised solar cell. You might have more success searching the internet using that term. Perhaps before looking at the Gratzel cell we should consider an 'ordinary' or solid state photovoltaic cell. A simple photovoltaic cell
When light strikes the top sheet (the p side) it reverses this tendency: light excites an electron out of a normal valence shell in the p layer into a delocalised state (as in metals): this electron can then flow into the other sheet of silicon (leaving a positive 'hole' behind) and so into an external circuit as in the diagram. Electrons can flow from the external circuit into the positive holes in the p layer. Current flows as long as light strikes the top surface. This is the common form of solar cell for directly collecting solar energy. It is commonly used in remotely-located signalling devices and also in solar powered calculators. There have been suggestions that the energy gain factor for the Gratzel cell may be greater than for the silicon-based photo cell. The energy gain factor of a device is the ratio of the amount of energy produced by the device during its useful lifetime to the amount of energy used to make the device. The following websites provide more
information: When transuranic elements were first being made, the usual practice was to let the team that had discovered the new element name it. However disputes arose when two groups claimed almost simultaneous discovery and hence naming rights. Hence a system of temporary naming was introduced until agreement could be reached on a permanent name. The temporary name was a Latin version of the atomic number with 'ium' stuck on the end with the symbol being the first letter of each of the digits in the name: unnilquadium, symbol Unq, was the early name for rutherdfordium (atomic number 104). The Latin words are
4. Stable and unstable nuclei and positron emission Figure 3.1 on page 75 shows the range of known stable and unstable nuclei. Exercise 6 on page 79 shows (1) that a beta emission decreases the n to p ratio (and so explains how isotopes in the blue region above the orange region convert back to stable ones) and (2) that an alpha emission increases the ratio (for isotopes with n/p greater than 1); this would explain how isotopes in the blue region below the orange zone but above the n = p line could convert to stable nuclei. However alpha emission only occurs with elements with high atomic numbers (that is with n/p > 1), so how do other unstable isotopes in the blue zone below the orange get back to stable ones? Often it is by the emission of what is called a positron, a particle of the same mass as an electron but with a positive electrical charge of the same magnitude as the charge on an electron. A positron is formed by a proton 'decomposing' into a neutron and a positron, symbol e+:
For example 18F ® 18O + e+ (9 protons give 8 protons) or 123I ® 123Te + e+ (53 protons give 52) Alternatively the symbol for a
positron is Positrons readily react with electrons to form gamma rays, the mass of the two particles being converted into energy according to Einstein's equation E = mc2. e+ + e– ® gamma rays (energy) There are no naturally
occurring positron-emitting isotopes. 070414 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||